发病机制
发病机制
发病机制:
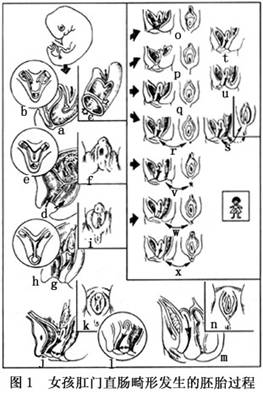
1.胚胎发生 在胚胎第3周末,后肠末端膨胀与前面的尿囊相交通,形成泄殖腔。中肾管(午菲管)―原肾管开口于泄殖腔中。泄殖腔的尾端被外胚层的一层上皮细胞膜所封闭,称为泄殖腔膜,使与体外相隔(图1a)。胚胎第4周,位于泄殖腔与后肠间的中胚层皱襞形成并向尾侧生长;同时,间充质于泄殖腔两侧壁的内方增生形成皱襞,向腔内生长(图1c)。它们构成尿直肠隔,将泄殖腔分为前后两部分,前者为尿生殖窦,后者为直肠,使两个系统的交通越来越小,逐渐形成一个小管道,称为泄殖腔管,于胚胎第7周时完全封闭。尿直肠隔由两个内胚层板构成(尿生殖层和直肠层),在两层之间充满中胚层组织和生殖胚芽。
尿直肠隔与泄殖腔膜的中央处融合,并向外突出成为会阴矩状突―未来会阴的胚芽。同时泄殖腔膜也被分为前后两部分,前者为尿生殖窦膜,后者为肛膜。胚胎第7~8周时,两个膜先后破裂(图1d)。肛门的出现不仅由于肛膜破裂,在此以前,从胚胎第5周开始,外胚层向肛膜的外表面发展,形成肛凹,肛凹逐渐加深接近肠管,肛膜破裂使起源于外胚层的肛凹与内胚层发生的直肠相通。
胚胎第4个月,会阴向前后方向迅速增长(图1j,l),因此使肛门后移至通常位置(图1m,n)。生殖器官和会阴的形成与上述过程同时进行。在女胎,内生殖器官由米勒管形成,该管开始与中肾管一起发展,向下延伸至中胚层的尿直肠隔的深部(图1b),米勒管的中段和下段靠近并融合在一起(图1e),形成子宫和阴道,其上部没有融合则形成输卵管(图1h),午菲管退化。
在女胎泄殖腔分隔以后,生殖皱襞的后半部与尿直肠隔的会阴矩状突愈合在一起(图1f),形成会阴和叉状的阴道前庭原基(图1i,k);生殖隆突没有愈合,变成大阴唇(图1f);生殖皱襞的前半部也没有愈合,形成小阴唇。
在女胎泄殖腔形成和分隔期受某种因素或致畸物质的影响出现发育障碍,可构成下列畸形(图1):O-直肠泄殖腔畸形;p-直肠膀胱瘘(米勒管中部未愈合时,这种畸形伴有双角子宫;下部未愈合时,伴有双阴道);q-
直肠阴道瘘;r-直肠前庭瘘;s-肛门正常,直肠前庭瘘;t-肛门直肠发育不全,无瘘;u-肛门发育不全,无瘘。
后期发育停止导致出生后病儿肛膜未破(v)。
会阴矩状突发育不全时,生殖皱襞是形成会阴的基本来源,生殖皱襞肥大,在通过肛管的正常肛穴部位愈合所致的畸形,称为隐蔽肛门(w)。
前会阴肛门(x)是会阴发育不良,肛门没有后移至正常位置的结果。
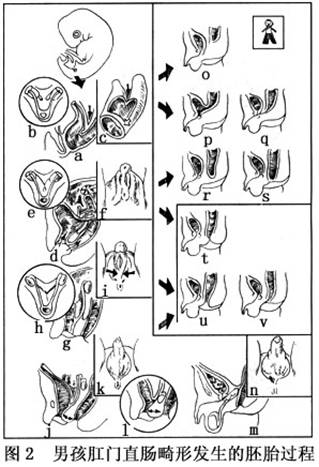
在没有分化性别期,泄殖腔的分隔过程在男胎(图2a,b)和女胎都一样,其基本差别是在内、外生殖器官和会阴形成时期出现的。午菲管发育成睾丸和中肾管变为输精管的同时,米勒管退化(图2e,g,h)。
在男胎形成会阴时,生殖结节增长形成阴茎。生殖皱襞左右愈合,覆盖于尿生殖窦的表面(图2f,i),形成前部尿道和尿道球部。在生殖皱襞外侧的生殖隆突则形成阴囊,沿矢状线愈合处为阴囊正中缝(图2k)。和女胎一样,男胎在第4个月以后的发育中会阴迅速向前后方向发展将肛门推移至正常位置(图2m,n)。
肛门直肠畸形的发生,男胎和女胎在原则上是相同的,只有解剖特点的区别。
泄殖腔分隔障碍的结果,是使尿生殖窦和直肠之间相通,在男孩可出现泄殖腔畸形,而较多见的是直肠泌尿系瘘,瘘管可位于膀胱三角部(图2p,直肠膀胱瘘)或尿道前列腺部(图2q,直肠尿道瘘)。当瘘管闭塞时出现肛门直肠发育不全,无瘘(图2o)。
胚胎发育后期出现发育障碍,结果可形成肛门发育不全,无瘘(图2r),肛膜未破(图2s),肛膜狭窄(图2t)。会阴发育不全,可构成前会阴肛门(图2u)和肛门皮肤瘘(图2v)、不完全性隐蔽肛门。
胎儿直至出生时直肠仍呈纺锤状,上端球状膨胀部称肛球,相当于成人的直肠壶腹部,纺锤状管以下另有一短而不明显的膨大部,称尾球,相当成人的直肠肛门部的下部。尾球存在的时间较短,第8周时大部已基本消失。肛门直肠正常的直肠闭锁,往往发生在肛球上端,相当于肛门上3~4cm处,可能与胚胎性狭窄有关。
会阴部肌肉是就地发育的,它起源于会阴部间质,在胚胎第2个月时已存在皮肌的形态,称泄殖腔括约肌。第3个月时皮肌分化为肛门外括约肌、肛提肌和尿生殖窦括约肌,当生殖器官形成后(第4、5月),尿生殖窦括约肌又分出膜部尿道括约肌、坐骨海绵体肌、会阴浅横肌等,以后再分出会阴深横肌。肛门直肠畸形病儿上述各肌虽然存在,但在高中位畸形时,外括约肌和肛提肌有不同程度的改变。
2.病理类型 先天性肛门直肠畸形的分类方法很多,名词术语也不统一,文献中对这些畸形的记载混乱,很难对比不同分类的治疗效果。
(1)Ladd-Gross分类法:过去,在我国多采用Ladd和Gross于1934年提出的4型分类法,即第1型肛门或直肠下端狭窄;第2型肛门膜状闭锁;第3型肛门闭锁,直肠盲端距皮肤有相当距离;第4型直肠闭锁。以后又将第3型分为高位和低位2型。这种分类方法是单纯从解剖形态上制定的,对手术方法和途径的选择以及预后的估计均无重要意义。
(2)国际分类法:1970年在澳大利亚召开的国际小儿外科医生会议上,制定了高位、中间位和低位的分类方法,它以该畸形的胚胎发生和病理改变为基础,对指导临床实践和估计预后均有帮助,是目前较合理的分类方法。该分类法是对许多分类方法的折中和修订,已被各国小儿外科医生广泛应用。
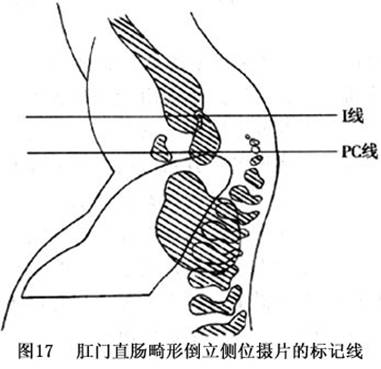
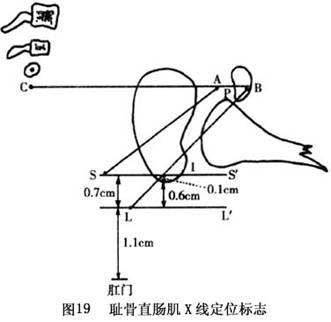
国际分类法的主要特点是以直肠盲端与肛提肌,特别是耻骨直肠肌的关系做为区分高、中、低位的标准,即直肠盲端终止于肛提肌之上者为高位畸形;直肠盲端位于耻骨直肠肌之中,被该肌所包绕为中间位畸形;穿过该肌者为低位畸形。Stephens发现在肛门直肠畸形病儿的耻骨直肠肌位置有改变,强调在做肛门成形术时注意保护该肌,并使直肠通过该肌环,对决定术后肛门排便功能有重要性。其次,国际分类提出了介于高低位之间的移行型,即中间位畸形,而这种畸形大部分应行骶-会阴肛门成形术,对合理选择术式有指导作用。
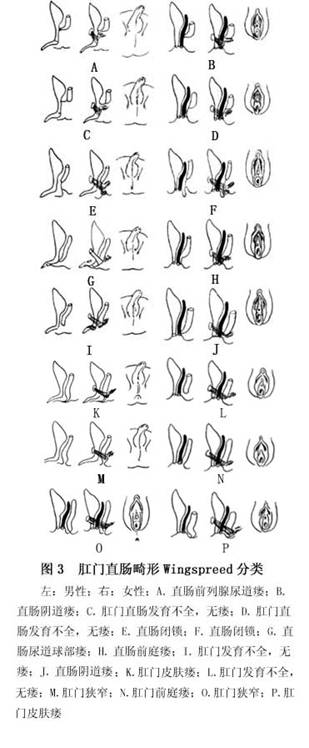
(3)Wingspread分类法:为便于应用1984年将国际分类法种加以简化,修改后的分类法称为Wingspread分类法,具体分类如下(表1,图3):
男性:
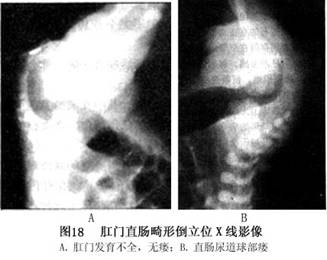
①高位:A.肛门直肠发育不全:a.直肠前列腺尿道瘘:瘘管开口于后尿道,无肛门内括约肌,外括约肌不明显,盲端位于PC线上。b.无瘘:盲端与尿道间可有纤维索带连接,无肛门内括约肌,仅有外括约肌痕迹,盲端平或高于PC线。 B.直肠闭锁:直肠盲端止于不同高度,肛门及肛管正常,有肛门内、外括约肌及肛提肌,且与肛管保持正常关系。
②中间位:A.直肠尿道球部瘘:直肠盲端位于尿道球部海绵体肌之上,耻骨直肠肌包绕直肠盲端瘘口,肛门内括约肌缺如,直肠盲端位于PC线与Ⅰ线之间。B.肛门发育不全,无瘘:直肠盲端终于尿道球部海绵体肌之上,耻骨直肠肌环绕直肠盲端。肛门内括约肌缺如,外括约肌仅见痕迹,直肠盲端位于PC线与Ⅰ线之间。
③低位:A.肛门皮肤瘘:瘘管开口于会阴部至阴囊缝线或阴茎腹侧的任何部位,以阴囊部居多。肛管呈瓣状,瘘管被菲薄的皮肤掩盖。耻骨直肠肌正常。B.肛门狭窄:肛门及内、外括约肌正常。
除上述3类畸形外,还有一些罕见畸形。
女性:
①高位:A.肛门直肠发育不全:a.
直肠阴道瘘:直肠盲端开口于阴道后壁中部。b.无瘘。B.直肠闭锁
②中间位:A.直肠前庭瘘:直肠盲端位于PC线上或稍下,瘘管长1~2cm,通过耻骨直肠肌,沿阴道后壁开口于阴道前庭部。B.
直肠阴道瘘:瘘管开口于处女膜上方,耻骨直肠肌环绕直肠盲端与瘘管。C.肛门发育不全,无瘘:直肠盲端终于阴道下端平面,尿道及阴道正常。直肠盲端位于Ⅰ线或其下。
③低位:A.肛门前庭瘘:瘘管甚短,直肠与阴道紧密相邻。耻骨直肠肌正常,有肛门内括约肌痕迹,肛门外括约肌有时存在,瘘口位于阴道前庭部,瘘口周围为黏膜。B.肛门皮肤瘘。C.肛门狭窄。
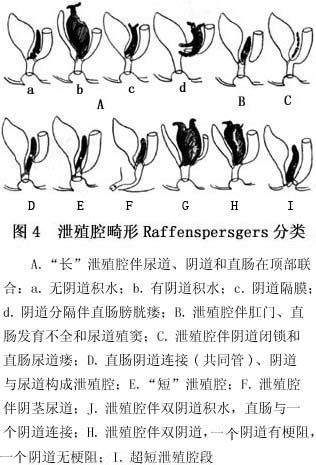
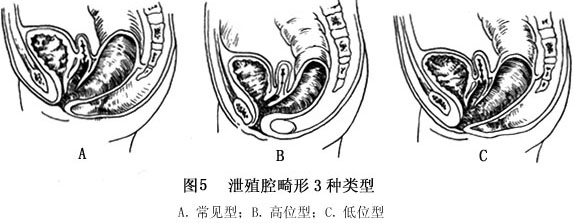
④泄殖腔畸形 这是一种较少见的肛门直肠畸形,即直肠、阴道、尿道共同开口在一个腔,一般按Raffenspersgers分型法分型(图4)。由于该分型法过于复杂,为便于应用,有人按病理解剖特点将其分为3种类型(图5)。
A.常见型:共同管长2~3cm,阴道大小正常,肌肉复合体及肛门外括约肌位置正常。
B.高位型:共同管长3~7cm,骶骨发育短小,肌肉发育薄弱,阴道狭小,骨盆前后径小,一般术后效果不理想。
C.低位型:共同管长0.5~1.5cm,又称低位
直肠阴道瘘合并女性
尿道下裂,盆部发育正常,预后较好。本病常合并双阴道、双子宫,约占60%,巨大阴道积液约占40%。
⑤罕见畸形
3.病理改变 20世纪70年代,不少学者对肛门直肠畸形病儿的盆腔结构进行解剖组织学研究,证明该畸形不仅肛门直肠本身有闭锁和发育不全,同时盆底肌内、骶骨、神经及肛周皮肤等均有不同程度的病理改变,肛门直肠畸形的位置越高,这种改变越明显,越严重。
(1)肌肉改变:
①耻骨直肠肌:Stephens对29例肛门直肠畸形病儿尸体进行解剖,发现2例高位肛门直肠畸形的男婴,耻骨直肠肌依附于尿道后壁;1例
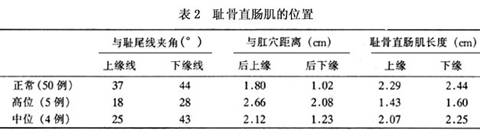
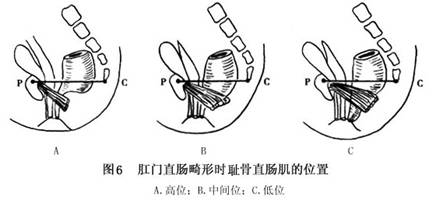
直肠阴道瘘者,该肌附着于阴道后壁并向前移位;而在患前庭瘘和肛门闭锁的病例中,该肌处于正常位置。Stephens指出耻骨直肠肌的发育与骶椎缺如有关,如骶2以下缺如,该肌不发育;骶3以下缺如,该肌发育薄弱;骶4以下缺如,该肌可正常发育。Kiesewetter曾做9例解剖,强调该肌有上移,即高位畸形时,耻骨直肠肌处于耻尾线(PC线)水平;而低位畸形该肌远离PC线。王常林观察和测量该类畸形病儿耻骨直肠肌的位置和长度(表2),发现高位畸形时耻骨直肠肌上、下缘延长线与耻尾线的交角明显小于正常儿。其后上、下缘与肛穴的距离明显大于正常儿,说明该肌上移,另外耻骨直肠肌的长度较正常儿短。耻骨直肠肌与外括约肌分离,与骶椎间隙增大,由脂肪占据。中位畸形时耻骨直肠肌虽有上移和缩短,但不如高位者明显,与正常儿比较无显著差异。该肌纤维包绕直肠盲端,且直肠盲端位置越低,被肌纤维包绕的越多。该肌在直肠盲端的后外方与外括约肌深浅部肌纤维相接。直肠前庭瘘者和低位畸形一样,耻骨直肠肌环绕于直肠或瘘道的后方,处于正常解剖位置。
总之,肛门直肠畸形病儿的肛提肌,包括耻骨直肠肌的发育良好,仅个别病例该肌缺如或发育不良。由于畸形类型不同,耻骨直肠肌的位置可发生改变,即高位畸形该肌明显向上向前移位,并短缩,呈闭锁状,依附于前列腺、尿道或阴道后方(图6),并与直肠盲端和外括约肌有一定距离。因此高位畸形行肛门成形术时,应设法使直肠准确地通过耻骨直肠肌环。中位畸形时直肠盲端位于耻骨直肠肌之中,被该肌所包绕,其肌纤维与外括约肌纤维相连。直肠前庭瘘和低位畸形耻骨直肠肌环绕于直肠后壁,基本处于正常位置。
②外括约肌:胚胎研究证明,外括约肌是单独发育的,与肛门直肠畸形的发生无关。Kiesewetter报道,肛门直肠畸形病儿存在外括约肌,在临床上有人用电刺激或肌电图研究观察,也证明了这一点。Smith在16例病儿中,经组织切片观察发现1例外括约肌缺如,另1例外括约肌前部缺如。Stephens也看到2例直肠尿道瘘的病儿无外括约肌。也有人认为高位畸形时外括约肌发育不良,或仅为痕迹器官。
王常林对16例肛门直肠畸形患儿的盆腔正中矢状断面标本进行解剖和组织学研究,证明外括约肌均存在,由于畸形类型不同,该肌的分布、形态、大小和肌纤维走行方向变化较大。用方格图表法对13例盆腔完整标本的外括约肌分布面积进行对比观察,并与6例正常新生儿标本对照,发现肛门直肠畸形病儿外括约肌的面积较正常儿增大,即低位畸形时外括约肌面积与正常儿基本一致;中位畸形时为正常儿的1.4倍;高位畸形时,仅1例外括约肌明显缩小,约为正常的1/2,并移位至尾骨尖部,其余均较正常儿的面积大,平均为正常儿的2.5倍,在外括约肌内部有不同程度的脂肪充填。
在正常儿盆腔正中矢状断面上,肉眼观察外括约肌呈前、后两团,位于肛管的前后方。肛门直肠畸形病例直肠盲端的位置越高,两团结构越不明显,不易分开,甚至失去正常形态。在镜下观察外括约肌纤维走行方向,正常儿外括约肌深浅部肌纤维呈横断面,低位畸形其肌纤维也为横断面;而中位畸形肌纤维多为斜行,仅少部分为横断面;高位畸形时,深浅部肌纤维多为斜行及纵行,呈横断面者甚少,有的病例几乎以纵行肌纤维为主,呈高柱状,总之外括约肌肌纤维走行方向异常紊乱(图7)。
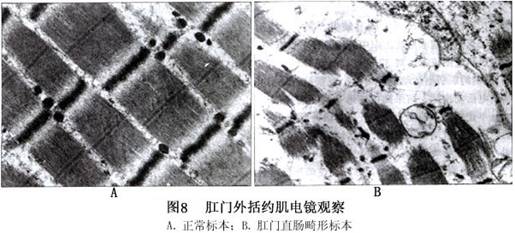
张志波等(1997)对肛门直肠畸形动物模型的肛门外括约肌进行组织化学观察发现,在高、中位畸形的单位面积中无论是肌纤维数,还是收缩,慢耐疲劳的Ⅰ型肌纤维所占比例均明显减少,而低位畸形则基本正常。我们对肛门直肠畸形肛门外括约肌的超微结构进行观察发现,部分肌原纤维排列紊乱,结构不清,有的呈溶解状态;Z带有不规则改变,扭曲、断裂;线粒体大小不等,嵴有缺失、断裂、空泡变,有早期髓鞘样变,这些改变可能与该畸形有骶髓和肛周组织中神经发育不良有关(图8)。
于明等采用体表电极肌电图技术,对32例肛门直肠畸形(高位7例,中位12例,低位13例)病儿肛门外括约肌肌电活动进行检测,观察肛穴处静止时波幅与频率及刺激时波幅与频率4项肌电活动指标,结果高位畸形有2项明显低于正常,中位有1项,而低位畸形肌电活动基本正常。从功能上证实肛门直肠畸形病儿无论高、中、低位畸形均存在肛门外括约肌,但直肠盲端位置越高,外括约肌发育越差。在肛穴的不同部位检测肌电活动结果表明,肛门直肠畸形儿肛门外括约肌肌电活动最强处不一定在正常肛穴位置,有11例(31%)位置偏前、偏后或偏侧,以高位畸形偏位者最多,中间位其次,低位又次之,各型又以向前偏位者多见。因此,对肛门直肠畸形病儿术前利用肌电图检查,确定肛门外括约肌的发育程度、位置及范围,术中尽量辨认和保存括约肌,不仅使直肠盲端通过耻骨直肠肌环,而且要穿过外括约肌中心是十分重要的。
③内括约肌:关于肛门直肠畸形病儿有无内括约肌,文献中说法不一。Stephens和Kiesewetter等认为肛门直肠畸形病儿无肛管,也无内括约肌。Scott(1959)在直肠前庭瘘病儿发现内括约肌存在。秋山洋(1973)和Nixon(1976)等证明,在低位肛门畸形时内括约肌存在。就是在1984年修定的肛门直肠畸形国际分类也标明:高位和中位畸形内括约肌缺如,低位畸形内括约肌存在。但早在1958年Bill就发现,伴泌尿生殖系瘘的高、中位肛门直肠畸形有内括约肌,并认为畸形的发生是直肠移行过程中受抑制而停滞于膀胱、尿道或阴道,未达到正常位置的结果。Gans(1961)对肛门直肠畸形进行病理研究,其结果与Bill的观察一致。王常林等(1983)报道了对10例肛门直肠畸形完整病理标本的组织学研究,在5例高位畸形标本中,3例于直肠远端肠壁环肌层有局限性增厚,范围较小;4例中位畸形中,3例也有局限性环肌增厚,范围稍大,多为前后两处,另1例前庭瘘,其内括约肌发育良好;1例低位畸形内括约肌基本正常。
故此他们认为肛门直肠畸形病儿内括约肌的发育程度与畸形类型有关,即位置越高,发育越差,甚至完全缺如。
1987年Lambrcht报道对33只新生仔猪肛门直肠畸形的形态学观察结果:公猪24只,母猪9只,高位畸形18只,低位畸形15只。所有动物均见有内括约肌环绕于瘘的近端,局限于瘘进入直肠盲端的开口部位。内括约肌的形态差异很大,有的像正常的内括约肌一样呈漏斗状;但大部分内括约肌分布较宽,呈圆盘状或平碟状。在肛门畸形瘘的近端有肛管的很多特征:A.被内括约肌环绕;B.在内括约肌部肠壁内神经节细胞减少或缺如;C.瘘的近端被有移行上皮;D.内有肛门腺。1990年Rintala对10例高、中位肛门直肠畸形病儿的直肠盲端、尿道瘘、会阴瘘进行组织学检查,发现9例有正常肛管移行上皮,在该区域内为低神经节细胞区,且胆碱酯酶呈强阳性反应。
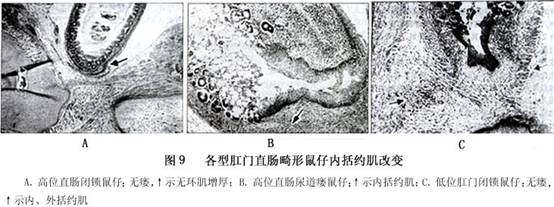
刘颖等(1996)对28只肛门直肠畸形鼠肛门内括约肌观察发现,有瘘型肛门直肠畸形其瘘管内覆有未角化的复层上皮,在该处直肠末端环肌层明显增厚,肌细胞发育正常。即有瘘型肛门直肠畸形具有明显的内括约肌,无瘘型肛门直肠畸形直肠盲端内无复层上皮,环肌也未增厚,即无内括约肌;而无瘘型低位畸形直肠盲端覆有角化鳞状上皮,范围较广,在有鳞状上皮的范围内环肌层局限性增厚明显,肌细胞发育正常,即存在内括约肌(图9)。
另外,也有人研究证明,在肛门直肠畸形病儿的瘘管处具有内括约肌功能。Ohama(1990)在术前由结肠造瘘口插入导管测量高中位畸形瘘管处的压力,当扩张直肠时,该处压力下降,即有正常肛管直肠反射。该反射反映肛门排便控制系统的完整性和神经肌肉的协调性,内括约肌的完整性是该反射存在的关键条件。
肛门内括约肌通常处于持续收缩状态,维持肛管高压,并构成肛管与直肠间的压力屏障,为控制排便的重要因素之一。有人认为内括约肌全部切除,肛管静止压力约下降50%。
上述资料说明,多数肛门直肠畸形(包括高、中位畸形在内)病儿都有内括约肌。因此,应行保留内括约肌的肛门成形术,即手术时保留直肠盲端及瘘管,这样可以最大限度地保存尽管是发育不全的内括约肌,以便获得较好的排便功能。自1982年以来李正等采用保留内括约肌的肛门成形术,对19例高位畸形病人术后随访6.5年,肛门功能临床和综合评定为优者分别由1982年前的23%、26.9%提高到63.2%和57.9%。Husberg 1992年报道对48例高中位肛门直肠畸形采用保留内括约肌的肛门成形术的随访结果,对4岁以上的22例进行直肠肛管测压,15例直肠肛管反射正常;其中11例排便功能正常,而6例该反射消失者均有污粪。
④肠壁纵肌:在部分高、中位畸形病例中,可见直肠盲端肠壁纵肌向下延伸,可延伸至外括约肌的肌纤维内,其长短不同。
(2)神经改变:
①骶髓改变:李龙等(1993)报道对10例肛门直肠畸形儿的骶髓标本进行观察,其中高位畸形4例,中位1例,低位5例。病儿末段骶髓均存在异常改变,其中6例标本的中央管呈菱形扩大,实质变薄;1例从第4骶髓节段以远,中央管和前正中裂未发育,左右前角内侧群的运动神经元在中线处融合;1例低位畸形末段中央管内有一矢状走行隔膜;另外2例,末段骶髓的中央管横向扩大,似脊髓裂样改变。
畸形标本中,有2例第4和第5及其以远的骶骨缺如,但是其相应的神经根却存在,并分别穿出硬脊膜。
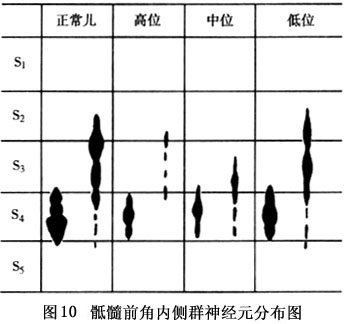
除上述形态改变外,先天性肛门直肠畸形儿骶髓前角内侧群的运动神经元的数目较正常儿减少,高、中位畸形和低位畸形分别为正常的34.4%和70.5%(图10)。
目前已明确,神经管为胚胎早期发育的中轴器官,它诱导附近各胚层结构的分化和发育。肛门直肠畸形儿末段骶髓的异常改变,可能意味着此畸形在胚胎早期,因尾端神经管发育异常,致盆底和会阴部组织发育畸形。
骶髓前角内侧群的运动神经元是盆底肌肉和肛门外括约肌的运动神经中枢,肛门直肠畸形儿此群运动神经元数目减少,与其周围神经的改变一致。
②骶神经改变:肛门直肠畸形病儿常伴有骶椎畸形。当骶椎椎体缺如时,可伴有骶神经的改变,缺如的节段越多,骶神经改变越明显。Smith解剖6例肛门直肠畸形儿尸体,2例第2骶椎以下缺如,未见骶神经及会阴神经;2例第3骶椎以下缺如,骶神经仅有3对,这4例骶神经缺如数目与骶骨缺如节段相一致,称为预料型。另2例第2骶椎以下缺如者,1例左侧没有骶神经,而右侧有7支骶神经发出,两侧会阴神经都来自于右侧骶神经;1例骶神经分别从第4、5腰椎之椎间孔、第5腰椎和第1骶椎之椎间孔发出,两侧会阴神经都存在。这种骶神经改变与骶骨缺如节段不一致,称为非预料型。
王常林等(1992)报道16具肛门直肠畸形儿尸体(高位畸形10例,中间位畸形6例)解剖发现:其中14例患儿解剖显示第2、3、4骶神经存在,该神经从相应椎孔发出后,沿椎体两侧下行,有分支到达盆底肌肉;在仅有3个骶椎的病例,硬脊膜终止于第3骶椎下方1.5cm处,在其最低处有一束马尾神经穿出,形成第4对骶神经;在第2、3、4椎体融合的病例,相当于第3椎间孔处发出一对骶神经。
骶神经与直肠盲端的关系:在10例高位畸形中,有5例直肠盲端位于第2骶椎水平以上,第2、3、4骶神经与直肠盲端无联系;其余5例,直肠盲端在第2骶椎水平以下者,1例第2骶神经进入直肠壁;3例第3、4骶神经,1例第2、3、4骶神经与直肠壁相连。中间位畸形6例,直肠盲端均与骶神经有联系,其中4例与第3、4骶神经相连,2例与第2、3、4骶神经相连。
肛门直肠畸形儿骶椎有明显改变者,可伴有骶神经的发育异常,直接影响本病的治疗和预后。
据Ленюшкин报道,在肛门直肠畸形术后排便功能障碍的病例中,约有10%的病例是骶椎畸形,神经发育障碍所致。因而临床上观察畸形儿骶椎改变具有重要意义。
③肛周组织中神经末梢改变:在正常儿盆底及肛周组织中共有4种感觉神经末梢存在:肌梭,位于耻骨直肠肌的前2/3段内和肛门外括约肌的中段内;环层小体,位于内括约肌与外括约肌之间的组织中和骶前间隙内;球样末梢,位于骶前间隙内;游离神经末梢,分布于肛管的黏膜上皮和肛周皮肤中。
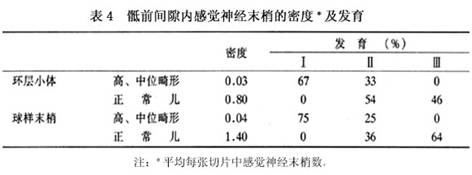
李龙等报道11例死于新生儿期的先天性肛门畸形儿(高位5例,中位5例,低位1例)盆底及肛周组织中的感觉神经末梢形态学观察结果,发现肌梭仅见于耻骨直肠肌中,分布在中1/3段内;在肛门外括约肌中未见肌梭(表3),且发育不良(表4)。
李正等发现,高位和中位肛门畸形儿耻骨直肠肌、肛门外括约肌和骶前间隙内的感觉神经末梢呈发育不良改变。一方面,感觉神经末梢的密度降低,以肛门外括约肌和骶前间隙内的感觉神经末梢降低为甚,同时尽管耻骨直肠肌中的肌梭数较正常儿少,但是确实存在有一定数量的肌梭;另一方面,骶前间隙内感觉神经末梢发育不良。
目前已经明确,肌梭、环层小体和球样末梢分别为牵张反射、压力感觉和温热感觉的感受器。许多学者认为,正常人耻骨直肠肌和肛门外括约肌中的肌梭是构成该肌肉在一般状态下持续收缩反射和扩张直肠时肛门外括约肌收缩反射的感受器,同时它与肛周组织中的环层小体、球样末梢、触觉小体等共同参与便意产生过程。
Kiesewetter在对高、中位肛门畸形儿术后复查时发现,刺激排便控制功能较好患儿的耻骨直肠肌区,可产生排便感。这可能是刺激兴奋了此肌肉中肌梭的结果。耻骨直肠肌中的肌梭是高、中位畸形儿重要的排便感受器。
正常新生儿耻骨直肠肌和肛门外括约肌中运动神经末梢的分布:运动终板分布于耻骨直肠肌的中段内和肛门外括约肌的两侧段内。两肌肉中,平均每张切片运动终板的数目分别为133.58±76.84和37.74±13.53。耻骨直肠肌和肛门外括约肌中,平均每张切片神经束的数目分别为94.06±45.43和66.16±32.82。 肛门直肠畸形儿耻骨直肠肌和肛门外括约肌中运动神经末梢的分布:运动终板的分布与正常儿相似,但是其面积较正常儿小,且着色淡,高位和中位肛门直肠畸形儿耻骨直肠肌中,平均每张切片运动终板的数目分别为14.00±11.30和18.31±8.38。肛门外括约肌中,运动终板数目分别为9.57±4.92和10.70±4.57。高、中位畸形儿两肌肉中运动终板的密度较正常儿明显降低。
高位和中位肛门畸形儿的耻骨直肠肌中,平均每张切片中神经束的数目分别为14.29±12.90和21.92±11.06;肛门外括约肌中,神经束的数目分别为9.29±7.13和14.67±7.93。高、中位肛门直肠畸形儿两肌肉中神经束的密度较正常儿明显降低。
运动神经末梢是控制肌肉活动的重要环节,高、中位肛门直肠畸形儿耻骨直肠肌和肛门外括约肌中的运动神经末梢呈发育不良改变,其程度与两肌肉中感觉神经末梢的改变一致。
④直肠远端肠壁内神经改变:肛门直肠畸形儿直肠远端肠壁内胆碱能、肽能和肾上腺能神经也有不同程度的改变。王伟等(1993)对8例无肛畸形儿(高位3例,中位3例,低位2例)直肠远端肠壁内胆碱能、肽能和肾上腺能神经分布进行观察发现:在黏膜下层AchE阳性神经丛及神经节细胞数及肌间的神经丛数较正常儿略有减少,酶活性减弱,而肌间的AchE阳性神经节细胞数则明显减少,并且以不成熟型为多,每个视野面积内仅为1.3个,而在正常儿则为2.7个;在黏膜下层和肌间,SP能阳性神经丛和神经节细胞数量也明显减少,新生儿位于黏膜下层及肌间的神经节细胞数为12.42个和13.28个,而肛门直肠畸形儿则仅为2.50个和7.83个。其免疫反应新生儿呈弱阳性者分别为15.9%和7.5%,而肛门直肠畸形儿则高达53.3%和100%。另外,在肌间肾上腺能阳性神经纤维较正常儿减少,荧光强度减弱。
⑤肛门部皮肤神经改变:正常儿肛门部皮肤有丰富的感觉神经末梢,能辨别直肠内容物的性质是固体、液体和气体。因此,许多学者强调行肛门成形术时应充分利用肛穴部的皮肤形成肛管,以保留感觉功能。
Kiesewetter研究肛门成形术后肛门直肠的感觉功能发现,低位畸形行会阴肛门成形术的病例肛门直肠感觉功能良好;而高位畸形行拖出型腹会阴肛门成形术的病例,仅在齿状线上1~2cm的直肠黏膜有感觉,其他部位无感觉,认为这种感觉的产生是皮肤感觉神经末梢进入直肠远端黏膜的结果。
Ленюшкин对肛门直肠畸形病例肛穴部皮肤进行组织学研究,发现该处皮肤菲薄,乳头变平,全部表皮被2~3层细胞和角化层覆盖,特别是没有神经纤维和神经末梢,像神经切除术后的皮肤组织学改变一样。肛穴部
皮肤发育不良的面积为(0.3~0.5)cm×(1.0~1.5)cm,其大小与肛门直肠畸形的位置高低无关。
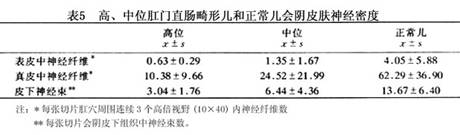
李正等对11例肛门直肠畸形儿肛门部皮肤进行组织学检查,结果与Ленюшкин的观察不一样,在该处皮肤与皮下组织中均有神经纤维存在,但是高位和中位畸形儿神经纤维的密度明显低于正常儿,且高位低于中位(表5)。
总之,从骶髓到盆腔和肛周组织中各种神经末梢的改变,即神经病理改变也是肛门直肠畸形的重要病理改变,其病理改变程度与畸形类型有关,畸形位置越高,其神经病理改变越明显。
各类肛门直肠畸形病儿盆腔结构改变举例如下。
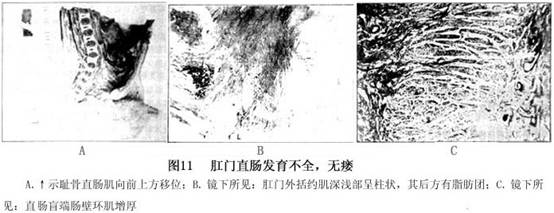
例1:男,高位肛门直肠发育不全、无瘘。在盆腔正中矢状断面上肉眼可见直肠盲端位于第1骶椎水平,距肛穴皮肤5.0cm,耻骨直肠肌明显向前上方移位、短缩,呈闭锁状态,依附于前列腺后方。该肌距直肠盲端2.0cm,并与外括约肌分离。外括约肌处于耻骨直肠肌和肛穴之间,其前后方有脂肪充填。
镜下所见:耻骨直肠肌未显示。外括约肌深浅部位于尾骨尖前下方的脂肪团块与前列腺之间,肌纤维基本为纵行走向,呈柱状直达肛穴,将外括约肌皮下部分为前后两部,该肌靠上方肌纤维排列致密,靠下方肌纤维排列较疏松,肌纤维间有脂肪和结缔组织充填。外括约肌皮下部面积较广,其肌纤维平行皮肤走向。直肠盲端镜检,肠壁部分环肌有增厚(图11)。
另1例高位肛门直肠发育不全,无瘘。在盆腔正中矢状断面上,肛穴上方被一巨大的梨形脂肪组织所占据,外括约肌明显向后上方移位,位于尾骨尖的前下方(图12),面积仅为正常外括约肌的1/2。在该肌中部有一条纤维脂肪隔将其分为前下和后上2部。前部与耻骨直肠肌相连,肌纤维呈斜断面及横断面;后部与尾骨相连,肌纤维多呈斜断面。
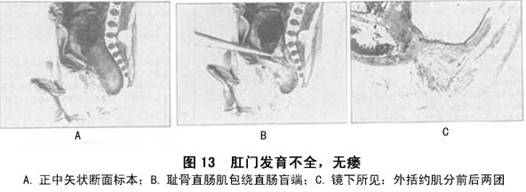
例2: 男,中位肛门发育不全,无瘘。在盆腔正中矢状断面上可见直肠盲端扩张,位于尾骨尖水平,并受2、3、4骶神经支配。耻骨直肠肌包绕着直肠盲端,较菲薄,外括约肌在耻骨直肠肌与肛穴之间。
镜下所见:耻骨直肠肌包绕于直肠盲端。外括约肌深浅部位于耻骨直肠肌下方,中间有脂肪及结缔组织将其分为前后2部(图13)。前部较小,位于海绵体肌后方,肌纤维多呈横断面和斜断面;后部较大,与耻骨直肠肌相连,多呈横断面、斜断面。
直肠盲端肠壁较薄,前壁有一处可见环肌增厚。
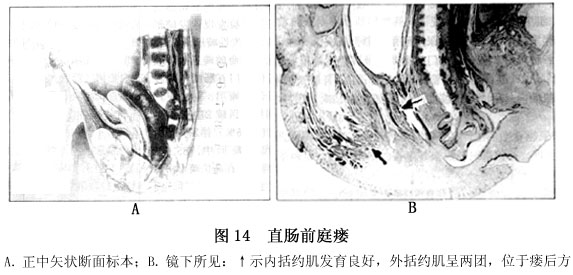
例 3: 女,直肠前庭瘘。在盆腔正中矢状断面上,直肠不扩张,受2、3、4骶神经支配。耻骨直肠肌绕过直肠及瘘道后方。外括约肌分前后两团,较易辨认,位于瘘道后方。骶骨有反曲,第2、3、4骶椎融合,只显示一个骨化核。
镜下所见:耻骨直肠肌环绕于直肠及瘘道后方,外括约肌呈前后两团,均位于瘘道后方。前团面积小,与瘘道后壁相邻,肌纤维呈横断面;后团面积较大,其上方与耻骨直肠肌相连,肌纤维多呈横断面,部分为斜断面,皮下部从尾骨尖向下,与皮肤平行走行到肛穴处中止。外括约肌的位置、结构基本正常(图14)。瘘道自直肠远端向下伸延,该处肠壁环肌已开始逐渐增厚,直达瘘道的远端,已形成较完善的内括约肌。瘘口周围结缔组织致密。
例 4:女,肛门皮肤瘘。在盆腔正中矢状断面上,瘘开口于阴唇后联合稍后方,直肠被一巨大多囊性肿物挤压向前移位,仅有2个骶椎,第2骶椎椎体明显变小(图15)。
镜下所见:外括约肌深浅部呈一团,与耻骨直肠肌相连,肌纤维多呈横断面,皮下部与皮肤平行走向,分布于瘘口后方。肿瘤病理诊断:骶前畸胎瘤。
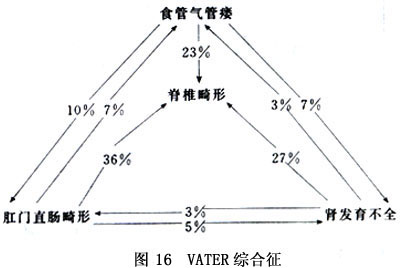
4.伴发崎形 先天性肛门直肠畸形经常伴发其他畸形,一般报道其发生率为28%~72%。Stephens和Smith在246例肛门直肠畸形中发现149例(60.6%)伴发1种或多种畸形。有人收集3223例肛门直肠畸形,伴发畸形的发生率为43.4%。但实际上比上述的数目要多,因为有一些内脏畸形尚未被发现。有人报道尸检发现伴发畸形为92%。有些病例为多发性畸形,约1/5病例伴发严重的危及生命的畸形。多数学者一致认为,高位肛门直肠畸形伴发畸形的发生率多于低位畸形,而且更严重。Cook报道在利物浦医院219例高中位畸形中,159例(72.6%)伴发其他畸形,而在165例低位畸形中,35.2%(58例)伴发其他畸形。在高位畸形中伴发畸形的发生率男女比例基本一样,而在低位畸形中伴发畸形的发生率女多于男,分别为50%和25%。高位畸形中76例(34.7%)死亡,而低位畸形仅20例死亡(12.1%)。二组中死亡病例多数死于其他系统畸形。伴发畸形最多的为泌尿生殖系畸形,其次为脊柱,特别是骶椎、消化道、心脏以及其他各种畸形。有人将肛门直肠畸形及其伴发畸形归纳为VATER综合征:(V:脊柱、心血管;A:肛门;T:气管;E:食管;R:肾脏及四肢),并指出某些畸形合并发生的非随机倾向,用图表示彼此间相对发生的几率(图16)。
肛门直肠畸形多伴发泌尿生殖畸形,且多为上尿路复合性严重畸形,近年来文献报道较多。目前不少医生在发现肛门直肠畸形之后,没有检查有无伴发泌尿系畸形,以致使大部分并发畸形被漏诊,或只到晚期已发生严重结构和功能障碍,发展到慢性肾功能不全的程度才确诊。
自1952年曾有学者报道120例肛门直肠畸形中有41例(34%)合并泌尿系异常后,文献中各学者报道的发生率差别较大,从19%~77%。1974年宫野等在100例肛门直肠畸形儿中60例进行了IVP检查,58例进行了排尿时膀胱尿道造影检查,发现上尿路异常32例(53%),下尿路异常16例(26%)。在平本等报道的273例肛门直肠畸形中,上尿路异常53例(19.4%),下尿路异常23例(8.8%)。一般上尿路畸形包括
单侧肾缺如、
肾发育不良、孤立游走肾、融合
异位肾、
马蹄肾、单或双侧
肾积水、巨输尿管、
膀胱输尿管反流等,以
单侧肾缺如、
肾发育不良和
膀胱输尿管反流较常见。下尿路畸形包括神经膀胱、
膀胱外翻、
尿道狭窄、
尿道下裂等。
Атакулов(1983)在186例肛门直肠畸形中发现有81例(44%)伴发泌尿生殖畸形,其中上尿路畸形48例,包括
肾发育不全、肾盂输尿管积水、双肾
双输尿管畸形、输尿管开口异位、
输尿管囊肿等;下尿路畸形53例,包括神经膀胱、直肠尿道瘘、
尿道狭窄、憩室和重复畸形等;生殖系统畸形16例,如
尿道下裂、泄殖腔畸形、
隐睾等。Атакулов最初只对有泌尿系症状的肛门直肠畸形病例进行泌尿系检查,结果在124例中发现34例(27%)泌尿系畸形,以后对肛门直肠畸形病例常规做泌尿系检查,在61例中发现47例(占77%)泌尿系畸形,较前期增加2倍。因此,他强调对每个病人都应常规进行泌尿系检查。同时还发现男孩伴泌尿系畸形者为女孩的2倍;高位畸形伴泌尿系畸形者占60%,而低位畸形仅占20%。
合并泌尿系异常的发生率与肛门直肠畸形类型有关,肛门直肠畸形的位置越高,合并泌尿系异常的可能性越大,且畸形严重。寺岛报道在102例肛门直肠畸形中约2/3高位、1/2中位和1/3低位合并泌尿系异常。
在合并泌尿系异常中,以
膀胱输尿管反流、
肾积水和肾缺如多见。在宫野的病例中19%(24例)为
膀胱输尿管反流,在高、中位肛门直肠畸形中占30%~40%,其中一半为重度返流,低位仅占8%。
Stephens报道两组病例,在30例直肠畸形中有13例伴发泌尿系畸形,28例肛门畸形中9例有泌尿系畸形,而两组中约有半数未做泌尿系检查,可见伴发泌尿系畸形者很多。
生殖系畸形也是肛门直肠畸形常见的伴发畸形,因为它们有共同的胚胎发生学基础,即均为午菲管发育异常所致。在男婴常见
尿道下裂、
隐睾等,Cook报道在253例男婴肛门直肠畸形中有14例伴
尿道下裂。在收集的1272例病儿中有39例合并
尿道下裂。另外也有伴发少见的
阴茎前阴囊畸形者。
在高位肛门直肠畸形的女婴中,内生殖器畸形也较常见,包括阴道缺如、双阴道、阴道闭锁、子宫阴道积液、子宫缺如、双角子宫等。据Hasse统计,在1272例肛门直肠畸形中有10例阴道缺如。
近年来中国医科大学附属医院无选择地对28例肛门直肠畸形病儿进行了静脉肾盂造影(IVP)等泌尿系检查,属高位肛门直肠畸形的15例,中位10例,低位3例。伴有各种瘘道者占71.4%,其中膀胱瘘1例,尿道瘘11例,泄殖腔瘘3例,阴道瘘2例,舟状窝瘘1例,会阴瘘2例;无瘘8例。发现伴有上尿路异常者10例(35.7%),其中右肾及输尿管缺如2例,左肾及输尿管缺如4例,右重肾及重输尿管1例,右
肾发育不良1例,双侧肾异位1例,膀胱输尿管返流1例,输尿管开口异位1例。共7种异常11个病变(其中1例同时患有双侧病变)。本组病例中以肾及输尿管缺如最多,占6例,发生在高位肛门直肠畸形者5例,发生在中位肛门直肠畸形者1例。
伴有泌尿系瘘以外的下尿路异常3例(10.7%),
膀胱外翻、
尿道上裂、
膀胱憩室及
尿道憩室各1例,共4种异常(其中1例同时伴有
膀胱外翻和
尿道上裂)。伴发其他各种畸形者17例(60.7%),其中有2种以上其他畸形者7例(25%),多见骨骼和生殖系异常、先天性心脏病、神经系统及消化道异常。在骨骼异常中以腰骶椎异常多见,有9例(32.1%),半椎体、腰椎融合各3例,
脊柱裂4例,骶骨发育不良及骶骨平直各1例;其次为多指、肋骨和骨盆多发畸形。生殖系异常可见
隐睾、双阴道及
隐匿阴茎等。先天性心脏病2例均为室间隔缺损。还可见直肠重复畸形及
大脑发育不全、会阴部脂肪瘤和腰骶部脊膜膨出等畸形。
肛门直肠畸形合并有泌尿系异常早期多无泌尿系症状,往往易被忽视。因不能及时诊断和处理,使很多泌尿系畸形不能得到矫治。一些重要的泌尿系异常如
肾积水、
膀胱输尿管反流等,由于未能及时诊断和治疗,易造成
尿路感染,甚至导致肾功能障碍。因此,对肛门直肠畸形,特别是高、中位畸形患儿常规进行泌尿系检查十分必要。一旦确诊合并重症
肾积水或
膀胱输尿管反流应及早矫治。
脊椎,特别是腰骶椎畸形也是肛门直肠畸形经常伴发的畸形,自Hohl于1852年首次报道肛门直肠畸形病儿伴发骶椎畸形以来,有关报道逐渐增多,但伴发腰骶椎畸形的发生率各作者报道不一,为2.5%~66%。腰骶椎畸形的发生率与肛门直肠畸形类型有关。
Stephens报道30例高位肛门直肠畸形,其中17例合并骶椎异常;而28例低位畸形中仅有2例合并骶椎异常。里村报道26例高位畸形中11例(41%)合并腰骶椎畸形,在25例低位畸形中7例(28%)合并腰骶椎畸形。有人报道,2/3的高位畸形儿(男、女均同)和1/3的男性低位畸形儿伴发骶骨畸形,而女性低位畸形儿则很少有骶椎异常。
李正等对113例肛门直肠畸形病儿(其中高位32例,中位40例,低位41例)腰骶椎正、侧位X线片进行研究,结果发现:X线片上腰骶椎完整者97例,其中有异常者52例(53.6%),其发生率明显高于对照组(9.5%)。高中低位畸形骶椎异常的发生率分别为66.6%、58.3%和40.5%,其中多发性异常则各为50%、25%和0.97例腰椎X线片上,11例(11.3%)有腰椎异常,即腰椎骶化5例,半椎体4例,腰椎融合、发育不全和旋转畸形各1例,共12例次。
在113例骶椎X线片上,57例(50.4%)有各种异常,共94例次。骶椎异常的发生率与畸形类型有关,高位畸形最多,为62.5%;中位55%;低位为36.6%。23例(20.4%)有2个以上多发性异常,最多者有3~4个,在高、中、低位畸形中多发性骶椎异常的发生率依次为40.6%、22.5%和2.4%。在骶椎异常中,骶椎缺如和发育不全最多,共41例次(36.2%),在X线片上有各种不同的表现,可以是全骶椎缺如,也可以是部分缺如,即有1个或2个以上椎体缺如;有的表现为椎体侧块缺如,多为一侧,也有两侧者;有的椎体发育较小,或同时有2~3个椎体融合在一起。其次骶骨平直或反曲,即骶骨正常弯曲消失,甚至远位骶骨向后方反曲,本组共30(26.5%)例次。隐性骶椎裂也较常见,共28(24.8%)例次,多发生在第1、2骶椎,个别的为全骶椎裂。有9例(8.0%)为骶椎腰化。
有些腰骶椎异常无临床症状,对患儿无任何影响,如一部分隐性腰骶椎裂、腰椎骶化和骶椎腰化等。正常人有隐性脊椎裂者为5%~6%。本组有28例肛门直肠畸形患儿有此改变,占24.8%,明显高于正常人。腰椎半椎体和旋转畸形可造成脊柱侧弯,随年龄增长而加重。骶椎缺如或发育不全与肛提肌和骶神经的发育有密切关系。有人报道,第4、5骶椎缺如者,肛提肌发育正常;第3骶椎以下缺如,该肌发育薄弱;而第1、2骶椎以下缺如,该肌不发育。一般骶椎缺如与骶神经缺如是一致的,第3骶椎以上缺如或发育不全可严重地累及肛提肌及其支配神经,导致术后肛门功能障碍,引起完全的或部分的大小便失禁。但也有少数病例,骶骨虽缺如,骶神经尚存在,其功能良好。有人报道的246例中,51例有骶骨畸形,其中12例有严重的功能障碍。在Ленюшкин报道的无肛畸形术后大小便失禁的106例患儿中,约10%(12例)的患儿伴有骶椎全部缺如或骶椎裂。可见严重的骶椎异常可影响预后。未见全骶椎缺如者,第3骶椎以下缺如或发育不全者18例,其中8例(44.4%)术后排便功能有明显障碍,无论是临床评分和(或)直肠肛管测压、肛门外括约肌肌电图和钡灌肠X线检查评分均较差,可能与骶骨异常有关。
李正等观察到伴骶骨平直或反曲者共30例(26.5%),其发生率与肛门畸形类型有关。骶骨平直或反曲与排便功能的关系尚不清楚。但其中16例单纯骶骨平直或反曲无其他改变的患儿中,有5例(31.3%)经临床和客观检查有排便功能障碍,值得进一步研究。
鉴于肛门直肠畸形伴发脊椎畸形的发生率很高,因此,对每个肛门直肠畸形病儿,特别是高、中位畸形者应做脊椎X线片检查,以便及早了解伴发畸形,对估计预后和及时采取治疗措施有益。
除脊柱畸形外,肛门直肠畸形伴发四肢骨骼畸形者也常有报道。有人报道肛门畸形病儿中有2%有桡骨发育不全,在有下肢畸形的病儿中更为常见。
肛门直肠畸形伴发心脏及大血管畸形者也较常见。在一组384例肛门直肠畸形中,有心脏和大血管畸形者39例,占10.2%。其中219例高、中位畸形中有30例心脏大血管畸形,165例低位畸形中有9例。法洛四联症和巨大室间隔缺损是最常遇到的畸形,其死亡率高,一般须急诊做心外科手术。近来有人指出,凡肛门直肠畸形手术经过顺利,而术后吸奶无力、气急及皮肤苍白者,经检查多伴有室间隔或房间隔缺损。
肛门直肠畸形还可伴发各种各样的消化道其他畸形,据Acgill报道约占10%,如该畸形伴食管闭锁的报道越来越多。在利物浦医院384例肛门畸形病儿中31例合并食管闭锁,占8.1%。伴发巨结肠的发生率说法不一,有人报道88例肛门直肠畸形中2例合并先天性巨结肠,占2.3%;而Kiesewetter等人收集35个医疗中心的病例,发现其发生率为3.4%;Santulli等人调查结果,在1166例肛门畸形中仅1例伴无神经节细胞症。我们近30年来治疗肛门直肠畸形1000余例,也只见到1例伴发巨结肠。肛门直肠畸形也可伴发肠闭锁、环状胰腺、肠重复畸形、肠旋转不良等畸形。因此,对肛门直肠畸形病儿腹部X线片上腹腔无气体者,应警惕消化道其他部位也有梗阻。另外肛门直肠畸形也可合并有罕见的多种畸形组合在一起的复杂畸形,如内脏外翻、膀胱小肠裂等。总之,肛门直肠畸形病儿同时可伴发其他脏器畸形,而且可以有几种畸形同时存在。有的伴发畸形可直接影响预后,甚至危及病儿生命。因此,对肛门直肠畸形病儿应进行全面检查,特别是对泌尿系和骶椎检查不容忽视,以免遗漏伴发畸形。
治疗
治疗:
1.按患病类型来分 先天性肛门直肠畸形的治疗方法,根据其类型及末端的高度不同而异。
(1)会阴前肛
门无狭窄、排便功能无障碍者:一般不需治疗。肛门或直肠下端轻度狭窄,一般采用扩张术多能恢复正常功能。扩张方法是用特制的金属探子,自肛门插入直肠内,最初1次/d,留置15~20min,逐渐改为隔天1次或每周1~2次。一般持续6个月左右,直到排便正常,且能保持狭窄不再复发为止。探子应由小到大,直到能通过食指为止。并应教会家长用手指进行扩肛。如肛门显著狭窄,须行手术治疗。
(2)低位肛门直肠畸形:包括有瘘和无瘘者,以及肛门闭锁伴前庭瘘者应行会阴肛门成形术。对无瘘或有瘘但不能维持排便者,一般需在生后1~2天内完成手术。对伴有较大瘘孔,如前庭瘘、肛门狭窄等,生后在一段时间内尚能维持正常排便,可于6个月左右施行手术。
会阴肛门成形术的方法是于正常肛门位置做“X”形切口,各长1.5cm,切开皮肤及皮下组织。用止血钳向深部钝性分离,找到直肠盲端,此时透过肠壁,可见深色胎粪。用组织钳钳住直肠盲端,或用0号丝线于直肠盲端缝合2针支持线,缝线仅穿过浆肌层,不要穿透肠壁全层,以免胎粪自针孔处外溢。用止血钳钳夹小纱布球,紧贴肠壁进行钝性分离,先游离直肠后壁及两侧壁,最后游离直肠前壁。前壁距尿道(或阴道)很近,为了防止损伤尿道(或阴道),于该处注入0.25%利多卡因溶液1~2ml,使肠壁与尿道(或阴道)壁分开,即可较易分离。游离直肠要充分,一般以使直肠盲端自然突出于皮肤切口之外0.5~1.0cm为宜。
用4号丝线于直肠壁前、后、左、右行浆肌层缝合4针,固定于括约肌。按“+”字形切开直肠盲端,排出胎粪。将皮肤切口的4个皮瓣尖插入直肠盲端“+”字形切口的间隙中,用1号丝线将直肠准确地与皮肤缝合,先在四角皮瓣的八个尖端缝合,然后在两缝线间再缝合1~2针,保留一条缝线以固定肛管。选适当粗的肛管,包以凡士林纱布,插入直肠内4~5cm。
肛门会阴瘘者,其直肠盲端与肛门皮肤的距离较近,多在1cm以内。于手术开始前,自瘘孔填入凡士林纱条,以防术中粪便外流。沿瘘孔两侧及后缘呈半环形切开皮肤,并于其中点向后方延长1.5cm。游离直肠后壁及两侧壁,前壁不需游离。待肠壁充分游离后,剪去已游离的部分瘘孔边缘,并沿瘘管纵行切开直肠后壁1~1.5cm,将直肠壁与括约肌缝合固定3针,结节缝合直肠与皮肤。
对较少见的阴囊或阴茎皮肤瘘,手术时不必游离和切除瘘管,仅于其基底部,即对入直肠的部分切断、结扎即可。此瘘管以后多发生机化而闭锁。如瘘管不能闭锁,于2~3岁时将其切除。
肛门前庭瘘者,在肛门正常位置作“X”形切口,以保存阴唇后联合的完整性。切开皮肤、皮下组织,向深部做钝性分离,以显露直肠盲端及瘘管。游离直肠后壁及两侧壁,于近前庭处先横断瘘管,再自下而上地将直肠前壁与阴道后壁分开(如先游离瘘管,再将其切断,因接近瘘管的直肠与阴道后壁紧密黏着,如勉强进行分离,则易造成阴道或直肠损伤)。然后将远端瘘管由前庭处的瘘孔向外翻出,并于靠近瘘管口处将其贯穿缝合结扎。缝合直肠与皮肤。
(3)中位肛门直肠畸形:常伴直肠尿道球部瘘或低位直肠阴道瘘等。因瘘管位置特殊,从盆腔或会阴部均不易暴露,应行骶会阴肛门成形术。此手术宜在患儿6个月左右施行,故对无瘘和伴直肠尿道瘘的中位畸形患儿,应先作横结肠造瘘,以解除梗阻症状。伴低位直肠阴道瘘者,其瘘孔较大,在一段时间内尚能维持正常排便,则不必作结肠造瘘。
骶会阴肛门成形术是于尾骨尖下方作半弧形切口,长约5cm,沿正中线切开肛尾肌膜,靠近中线向深部分离,以免损伤支配肛提肌的神经。中位畸形耻骨直肠肌包绕于瘘管及直肠盲端的后下方,用直角钳紧贴直肠做钝性分离,边分离边向前推进,张开两钳叶,直至钳尖插入肌环。动作要轻柔,以免撕断肌纤维。
在肛门处作“X”形切口,于外括约肌中插入止血钳,并轻柔地向上分离,使之与自骶部切口插入的直角钳相遇。然后将一条胶皮带穿过外括约肌中心及耻骨直肠肌环从两切口引出作牵引用,用宫颈扩张器逐渐将两肌环扩大至能通过直肠为止。
对伴有尿道或阴道瘘者,应在直视下游离瘘管,并将其切断、缝扎或缝合残端。充分游离直肠,使直肠无张力地能自然下降到肛门切口为止。从肛门切口插入组织钳夹住直肠盲端,将其缓慢地牵至肛门。直肠与皮肤用丝线缝合。伴尿道瘘者应作耻骨上膀胱造瘘,并取出尿道内导尿管。
(4)高位肛门直肠畸形:包括无瘘和有瘘以及直肠闭锁的病例。确定诊断后,为了挽救患儿的生命,应作横结肠或乙状结肠造瘘术,以解除梗阻症状。待6个月后,再行骶腹会阴肛门成形术。
在尾骨尖下方横行切开皮肤2~3cm,沿中线切开肛尾肌膜,并向深部分离。高位畸形时耻骨直肠肌向前上方移位,位于尿道或阴道壁后方。显露该肌后,用直角钳紧贴尿道或阴道后壁,边张开两钳叶进行分离,边向前推进,直至钳尖插入肌环。然后将直角钳尖端向后至会阴部新肛门处,作会阴部切口,使之与骶部切口相通,并将一胶皮带穿过外括约肌中心及耻骨直肠肌环自两切口引出。
开腹游离直肠,沿直肠周围向下作钝性分离,显露直肠盲端。如有瘘管,应将其充分显露并钳夹、切断。断端用
碘酊、酒精处理后,丝线作贯穿缝合、结扎。同时应剥除残端遗留的黏膜,以免分泌的黏液积聚。充分游离直肠、乙状结肠,使其能无张力地达到会阴切口之外。用组织钳通过会阴部切口进入腹腔,钳住直肠并向下牵引直肠盲端至会阴部切口之外。在牵引时,防止发生扭转。抽出胶皮带,结节缝合直肠与皮肤形成肛门。
骶腹会阴黏膜下切除肛门成形术:骶及会阴部手术步骤与Stephens骶会阴肛门成形术相同。腹部手术,在盆腔腹膜返折处切开盆底腹膜,游离乙状结肠。在腹膜返折部位的直肠浆肌层与黏膜间注入生理盐水,使黏膜与肌层分离。环行切开浆肌层,保持黏膜完整。沿黏膜下层用锐性及钝性向远端分离直至直肠盲端后,结扎、切断瘘管,并切开肌鞘下端。然后用一把大弯血管钳通过会阴切口穿过肛门外括约肌中心、耻骨直肠肌及直肠盲端切口,钳夹直肠近端,自会阴部切口拖出形成肛门。此手术避免了盆腔剥离面广,损伤大的缺点,但有形成直肠肌鞘内积液、积脓以及尿道憩室的可能。
2.手术方法
(1)后矢状入路肛门直肠成形术:1980年由Vires和Pena提出的,适宜于高、中位肛门直肠畸形。
自骶尾关节上方到肛穴前方正中线上用针形电刀切开皮肤、皮下组织。在电刺激下观察两侧肌肉的发育情况,并从正中将横纹肌复合体分为左、右2部分,显露直肠盲端。先游离直肠后壁及两侧壁,最后游离直肠前壁。如有尿道(阴道)瘘,于直肠盲端缝支持线,切开肠腔,直肠前壁中央凹陷处即为瘘口。在直视下距瘘口3mm处切开肠壁一圈,用4-O~6-O尼龙无损伤针线缝合闭锁瘘口,并自下而上游离直肠前壁,直到直肠在无张力的情况下达到肛门处为止。如果直肠达不到肛门处或有张力,可将直肠周围纤维膜牵拉到紧张处,作多个不同水平的小横切口使之松解,可延长直肠约3~5cm,或开腹游离直肠。如直肠盲端极度扩张,难以通过肌肉复合体时,应将直肠后壁作倒“V”形剪裁,使其直径为1.2~1.5cm。直肠置于左右2部分横纹肌复合体之间,将肌肉复合体与肠壁缝合固定数针,缝合修复肌肉复合体及外括约肌。直肠与肛周皮肤缝合形成肛门。
本手术的优点是所有操作都在直视下进行,术野清晰,避免了盲目地切开、分离,将手术损伤减少到最小程度。尽量保留直肠及肛周组织,恢复直肠与其周围组织的正常解剖关系,以便术后获得较好的肛门控制功能。
(2)泄殖腔畸形修复术:对泄殖腔畸形应于出生后立即作结肠造瘘,使粪流改道,保持泄殖腔出口清洁,防止发生尿路感染。根治手术的时间应根据患儿情况、畸形复杂程度及术者的经验而定,一般以6个月以后手术为宜。也有人主张阴道成形术应在青春前期完成。
术前应从泄殖腔开口做逆行造影,以了解畸形类型是常见型、高位型或低位型。不但要了解泄殖腔的大小、尿道瘘和直肠瘘的高度,还要了解子宫的发育情况和有无畸形,以便选择术式。
手术取后正中矢状切口。从骶骨中段到泄殖腔外口处,在电刺激引导下,在中线上分开外括约肌和肛提肌。充分游离泄殖腔管,显露直肠进入泄殖腔的入口。在该处直肠黏膜缝数根牵引线,于直肠和阴道共壁之间做黏膜下分离。一般分离到距阴道开口以上2cm,直肠与阴道壁开始独立分开,分离直肠的长度直至能无张力地达到肛门皮肤为止。直肠分开后可显露阴道后壁。用同样的方法将阴道从尿道与阴道的共壁间做黏膜下分离。此处分离比较困难,因为阴道从后面包绕尿道一半以上,而且组织弹性差。阴道分离后往往出现阴道前壁缺血。阴道分离得越长,缺血越严重,越易出现尿道阴道瘘。阴道游离充分后修复尿道,特别是共同管两侧横纹肌对控制排尿有重要作用。围绕着事先置入膀胱的尿管修复尿道,缝合两层,然后将阴道在尿道后方缝合于皮肤上。对分离时严重损伤阴道前壁的病例,为防止出现尿道阴道瘘,应将阴道扭转90°,即使有血循环的阴道侧壁接触尿道缝线。如阴道不能达到会阴皮肤,可选用下列方法作阴道成形:
①皮肤阴道成形术:适用于阴道缺损较短的病例。皮瓣从未来阴道部位的两侧皮肤或阴唇皮肤形成,应为保留皮下组织具有良好血液供应的全厚皮瓣,两侧皮肤缺损缝合闭合。
②肠管阴道成形术:阴道缺损较多或无阴道的病例,采用带肠系膜的回肠或乙状结肠修复阴道。即在尿道修复和直肠游离之后,开腹并切取一段带肠系膜的肠管,自会阴拖出。肠管近端缝合于子宫或阴道下缘,肠管远端缝合在会阴部皮肤上。
最后作直肠修复形成肛门,即将直肠置于肛提肌与外括约肌中心,并将肌肉与肠壁缝合固定数针,同时重建会阴体。
泄殖腔畸形修复术均需做耻骨上膀胱造瘘术。术后2周伤口愈合后,应扩张肛门及阴道。新阴道不能随身体发育而成比例的扩大,因此,阴道扩张要持续到青春期。